Unleashed Carcinogenic Molecules: How the EGFR Signaling Pathway Drives Tumor Growth and Becomes a Target for Precision Therapy

Unleashed Carcinogenic Molecules: How the EGFR Signaling Pathway Drives Tumor Growth and Becomes a Target for Precision Therapy
In the intricate kingdom of cells, every molecule operates under established rules. However, when the epidermal growth factor receptor (EGFR), a key molecule, becomes dysregulated due to some reason, overexpressed, and persistently activated, a "rebellion" that triggers uncontrolled cell proliferation begins quietly. As a central driver of tumor initiation and progression, abnormal activation of the EGFR pathway not only initiates the malignant transformation of cancer cells but also serves as a significant breakthrough in modern precision tumor therapy.
I. The Dual Faces of Tumor Genes: How Proto-oncogenes Become "Traitors"
Tumorigenesis arises from imbalances in the genetic world, with the interplay between proto-oncogenes and tumor suppressor genes being crucial:
Activation and Transformation of Proto-oncogenes: Proto-oncogenes (such as EGFR), originally responsible for regulating cell growth, undergo mechanisms like gene mutations, gene amplification, or chromosomal rearrangements, transforming from "gentle regulators" into "uncontrollable instigators." For example, EGFR gene amplification leads to overexpression of the receptor protein, continuously transmitting proliferation signals to the cell.
Five Categories of Oncogenes: Including growth factors (such as EGF), growth factor receptors (such as EGFR), signal transduction factors (such as HER2), transcription factors, and cell program death regulatory factors, these collectively form a molecular network driving tumors.
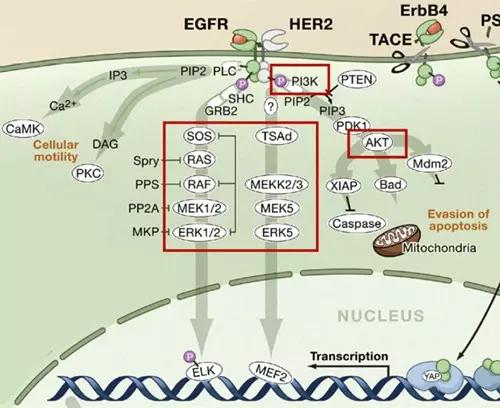
II. EGFR Pathway: The "Perpetual Motion" Switch for Cancer Cells
As a core member of the transmembrane glycoprotein tyrosine kinase family, EGFR (HER-1), along with HER2, HER3, and HER4, constitutes a key hub regulating cell growth:
(I) Cascade Reactions of Pathway Activation
Upon binding of EGFR to epidermal growth factor (EGF), dimerization occurs, activating intracellular tyrosine kinase activity and triggering three major downstream signaling pathways:
Ras-Raf-MAPK Pathway: Activates ERK1/2 kinases, promoting cell proliferation and differentiation, the core driving force behind cancer cell infinite division.
PI3K-AKT Pathway: Provides survival advantages and energy reserves for cancer cells by inhibiting apoptosis and promoting glucose metabolism.
JAK-STAT Pathway: Induces transcription of pro-growth genes, reinforcing the malignant transformation process of cells.
(II) Aberrant Expression and Consequences in Tumors
EGFR is highly expressed in various solid tumors: lung cancer (40%-80%), breast cancer (14%-91%), colorectal cancer (25%-77%), etc. Its overexpression or mutations (such as exon 19 deletions, L858R point mutations) lead to continuous activation of signaling pathways, driving cancer cell invasion, metastasis, and treatment resistance.
III. Targeting the EGFR Pathway: Precision Strikes from Mechanism to Clinic
In response to EGFR pathway abnormalities, scientists have developed various targeted drugs, ushering in the era of precision tumor therapy:
(I) EGFR-Targeted Drugs
Small Molecule Tyrosine Kinase Inhibitors (TKIs): Such as gefitinib and erlotinib, bind to the EGFR intracellular kinase domain, blocking phosphorylation signal transduction. Suitable for non-small cell lung cancer (NSCLC) with EGFR-sensitive mutations (exons 18, 19, 21), with an objective response rate of up to 80%.
Monoclonal Antibodies (mAbs): Cetuximab and panitumumab target the EGFR extracellular domain, blocking ligand binding and receptor activation, with significant efficacy in colorectal and head and neck cancers.
Novel Strategies: RNAi technology to degrade EGFR mRNA, antibody-drug conjugates (ADCs) for precise delivery of cytotoxins, and inhibition of receptor dimerization expand therapeutic boundaries.
(II) Downstream Key Targets: Challenges of KRAS and BRAF
KRAS Mutations: As a core signal transduction factor in the EGFR pathway, KRAS mutations at codons 12 and 13 (such as G12D) lead to persistent RAS protein activation, a major cause of EGFR-TKI resistance. Recent covalent inhibitors targeting KRAS G12C mutations (such as Sotorasib) have overcome the "undruggable" dilemma.
BRAF V600E Mutation: Common in melanoma (50%) and colorectal cancer (15%), its activation of the MAPK pathway has prompted the development of BRAF inhibitors like vemurafenib and dabrafenib, with combined MEK inhibitors significantly prolonging patient survival.
IV. Precision Screening and Resistance Management in Clinical Practice
Molecular Testing First: NCCN guidelines mandate testing for KRAS, NRAS, BRAF, and other gene mutation status before EGFR-targeted therapy. For example, KRAS wild-type patients respond better to cetuximab, while those with BRAF V600E mutations should avoid anti-EGFR monotherapy.
Exploration of Resistance Mechanisms: EGFR T790M mutations (accounting for approximately 50% of resistance cases), MET gene amplification, and HER3 bypass activation drive resistance. Third-generation TKI drugs like osimertinib target T790M mutations precisely, becoming the standard for second-line treatment.
From cellular signaling dysregulation to molecular targeted interventions, the research journey of the EGFR pathway has witnessed the transition of tumor therapy from "indiscriminate attack" to "precision guidance." With breakthroughs in downstream targets like KRAS and BRAF, and the emergence of novel drugs like bispecific antibodies and ADCs, the treatment landscape for EGFR pathway-related tumors is continually evolving. In the future, combining liquid biopsy and real-time monitoring of resistance mutations holds promise for achieving the ultimate goal of "individualized precision medication," ultimately deciphering the "code of(crazy proliferation code)" (translated as "code of uncontrolled proliferation") of cancer cells for humanity.



