Chromatin Immunoprecipitation (ChIP) Handbook

1. What is CHIP and what does CHIP study?
CHIP, the full name of chromatin immunoprecipitation, is an experimental technique used to study the interaction between protein and DNA. It can be used to detect specific protein-DNA interactions, such as histones, transcription factors, etc., binding to specific regions of the genome. ChIP technology can help researchers understand gene expression and regulation mechanisms in cells or tissues.
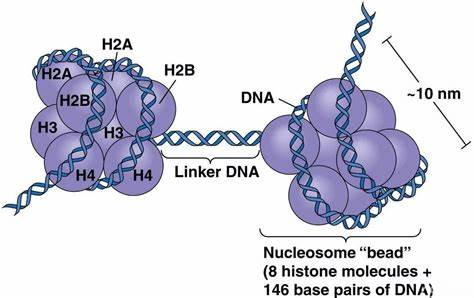
Figure 1 Histone octamer
* Noun explanation
Histone: A basic protein in the nucleus of eukaryotes. Their main function is to help compress and organize DNA molecules so that they can fit into the limited space within the nucleus. Together with DNA, histones form the basic building blocks of chromosomes, called nucleosomes.
Transcription factors: are a class of proteins that can bind to specific sequences on DNA molecules to regulate the transcription process of genes. Transcription factors play a vital role in cells. They control the time, place and extent of gene expression, thus affecting the function of cells and the development of organisms.
Transcription cofactors: They are a class of proteins that transmit regulatory information between enhancers and promoters. They are the core effector molecules of transcription activation and gene expression. Transcription cofactors enhance or inhibit the transcription of genes by interacting with specific transcription factors. They can show specificity for different types of core promoters, which means that different transcription cofactors may have preferences for different promoter types, thus affecting the specificity and diversity of gene expression.
2. What can ChIP do and what are the usage scenarios?
Determining the binding sites of specific proteins in the genome: By using antibodies directed against specific proteins, these proteins and the DNA fragments to which they bind can be precipitated from cells and then analyzed by sequencing or PCR to determine the DNA binding sites of the proteins.
Investigation of dynamic changes in protein-DNA interactions: ChIP experiments can be performed at different time points or under different physiological conditions to study changes in protein-DNA interactions, such as when cells differentiate, develop, or respond to environmental signals.
Analyzing histone modifications: ChIP experiments can be used to study the distribution in the genome of specific histone modifications that are associated with the activation or silencing of genes.
To study the DNA binding characteristics of transcription factors: Through ChIP experiment, the binding of transcription factors in the promoter region or enhancer region of genes can be determined, thus revealing its mechanism of regulating gene expression.
Whole genome analysis: Combined with next-generation sequencing technology (ChIP-seq), ChIP experiments can be used for genome-wide protein-DNA interaction analysis, providing high-resolution genome binding maps.
Examples of usage scenarios:
If you are interested in how a specific transcription factor regulates gene expression at different stages of development. You can run ChIP experiments at different stages of embryonic development, using antibodies against that transcription factor to precipitate the DNA fragments that bind to it. By sequencing these DNA fragments, you can determine the binding sites of this transcription factor in the genome and analyze the changes in these sites at different stages of development. These data can help you understand how this transcription factor regulates the expression of specific genes, thus affecting the development process of embryos.
Another example is the study of histone modifications. If you want to understand how the acetylation modification of histone H3 changes in the cellular stress response, you can do ChIP experiments before and after stress, using antibodies against acetylated H3. By comparing ChIP results before and after stress, you can reveal how stress affects histone modification patterns, which in turn regulate gene expression and cell function.
3. What are the types of ChIP?
Depending on whether or not a crosslinking agent is used, it can be classified into native ChIP (N-ChIP) and crosslinked ChIP (X-ChIP).
Standard ChIP (X-ChIP): In this experiment, a cross-linking agent (such as formaldehyde) is used to fix cells, which can fix the interaction between protein and DNA. Cross-linking is a necessary step because it stabilizes temporary or weak interactions between protein and DNA so that these interactions do not dissociate during subsequent chromatin preparation and immunoprecipitation. It is suitable for studying proteins that are not very stable to DNA binding, such as transcription factors;
Native ChIP (N-ChIP): In this experiment, no crosslinking agent was used. This method is commonly used to study those proteins that bind very stably to DNA, such as certain histone modifications, whose binding is stable enough that no additional cross-linking is required to remain bound to DNA during experiments.
4. What is the difference between magnetic bead method and agarose bead method in ChIP experiment?
Convenience of operation: The use of magnetic beads can quickly separate protein-DNA complexes, no centrifugation is required, easy to operate, the sample is not easily lost, and washes more thoroughly. The traditional method is to use agarose beads, which are inexpensive, but require centrifugation, take a long time, and the beads may break during centrifugation.
Specificity: The magnetic bead method usually has high specificity because the surface of the magnetic bead can bind the antibody more uniformly, reducing non-specific binding. The agarose bead method requires an additional pre-scavenging step to reduce this non-specific binding.
Cost: Although the magnetic bead method is easy to operate, the cost of magnetic beads is usually higher than that of agarose beads. The agarose bead method is less costly and suitable for labs with limited budgets.
Application scope: The magnetic bead method is suitable for high-throughput sequencing, such as ChIP-Seq, because the magnetic beads will not interfere during the sequencing process.
Agarose bead method: Suitable for traditional ChIP experiments, but may be limited in high-throughput sequencing.
* Why does the agarose bead method affect sequencing?
Binding of agarose beads to DNA: In ChIP experiments, the goal is to precipitate DNA bound to specific proteins, such as histone modifications or transcription factors. The surface of the agarose beads usually has protein A or protein G bound by covalent bonds, which are capable of binding to the Fc segment of the antibody. When an antibody binds to a protein, the DNA attached to it should also be captured. However, agarose beads themselves may also bind DNA non-specifically, and this binding may form a DNA encapsulation on the surface of the beads, thus hindering DNA elution and subsequent analysis.
Physical barrier: DNA that is non-specifically bound by agarose beads may form a physical barrier that prevents chemical components in the elution buffer from reaching all trapped DNA, or prevents enzymes and reagents of the sequencing platform from reaching target DNA. This physical barrier can affect the elution efficiency of DNA and the coverage of sequencing.
Elution efficiency: In the final stage of the ChIP experiment, the precipitated DNA needs to be eluted from the agarose beads for subsequent analysis, such as PCR, qPCR or sequencing. If the DNA is blocked, the elution efficiency will be reduced, resulting in a decrease in the amount of DNA eluted, which will affect the sensitivity and accuracy of subsequent experiments.
Sequencing coverage: Due to the DNA blocking phenomenon, some DNA fragments may not be effectively eluted and amplified, resulting in insufficient coverage of these regions during sequencing. This may lead to bias in the analysis of DNA-protein interactions, as certain regions may be mistakenly assumed that no interaction has occurred.
Sequencing accuracy: DNA blocking may also lead to a decrease in the accuracy of sequencing data. Since blocked DNA fragments may not be accurately read by the sequencing platform, this may lead to false negative results in the data.
5. How does ChIP work?
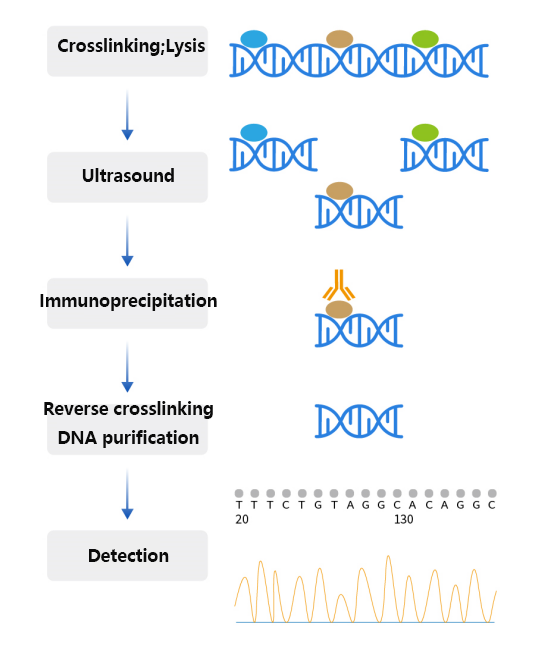
Figure 2 chip experiment flow
Taking cross-linked ChIP, that is, X-ChIP, as an example, the working principle of the experiment can be summarized as the following steps:
Cross-linking: Cells or tissues are first treated with formaldehyde or other cross-linking agents, a step that allows protein-DNA interactions and protein-to-protein interactions to be immobilized, preventing dissociation in subsequent steps.
Cell lysis and chromatin shearing: Cross-linked cells are lysed, releasing chromatin.
The chromatin is then sheared into smaller fragments, which can be achieved by sonication or enzymatic digestion, such as using the micrococcal nuclease MNase. This step is to shorten the target DNA fragment to facilitate subsequent precipitation and analysis.
Immunoprecipitation: Immunoprecipitation is performed using antibodies directed against specific proteins. These proteins may be transcription factors that bind directly to DNA, or histones or modified versions thereof that interact with DNA.
After the antibody binds to the protein of interest, these complexes are precipitated by beads coupled to protein A or protein G.
Reverse crosslinking: The precipitated protein-DNA complex is treated with heat or other methods to reverse the crosslinking and separate the protein from the DNA.
DNA purification and analysis: The released DNA is purified and analyzed by methods such as PCR, qPCR, microarrays, or sequencing (e.g. ChIP-seq) to determine the distribution of binding sites or histone modifications for specific proteins in the genome.
Data analysis: For high-throughput sequencing data such as ChIP-seq, analysis by bioinformatics tools is required to identify enriched DNA regions and reveal differences in protein-DNA interactions by comparing data from different samples or conditions.
6. How ChIP realizes cell cross-linking
In ChIP experiments, cell crosslinking is usually achieved by using formaldehyde or paraformaldehyde. The purpose of crosslinking is to immobilize the interaction between protein and DNA for analysis in subsequent experimental steps.
Formaldehyde: It can penetrate cells and react with amino acid residues of proteins and bases of DNA to form stable covalent bonds. The characteristic of formaldehyde crosslinking is that the covalent bonds formed are reversible, which means that the crosslinking can be terminated by specific chemical treatments (such as the addition of glycine) in subsequent experiments for DNA and protein separation and analysis.
Paraformaldehyde: Paraformaldehyde is a polymerized form of formaldehyde, usually in a powdered state, which needs to be dissolved in hot water to form a formaldehyde solution before use. Paraformaldehyde slowly releases formaldehyde in aqueous solution, so it can also be used for cross-linking of cells.
The advantage of paraformaldehyde is that it is more stable than formaldehyde, which makes it easier to store and handle, but it releases formaldehyde at a slower rate and needs to ensure complete hydrolysis into formaldehyde when used.
7. Effect of excessive crosslinking
Protein structure disruption: Excessive cross-linking may alter the three-dimensional structure of the protein, leading to protein denaturation, which affects its interaction with DNA.
Epitope masking: The cross-linking agent may react with the epitopes of the protein, thereby masking these epitopes, making it impossible for subsequent antibodies to effectively recognize and bind the target protein.
DNA damage: Excessive cross-linking may lead to excessive cross-linking between DNA strands, making it difficult for DNA fragments to be effectively cut during ultrasonic or enzymatic digestion, affecting the quality of chromatin fragmentation.
Decreased resolution: In ChIP experiments, the ideal DNA fragment size is typically between 200-800 bp. Excessive crosslinking may make it difficult for sonication to cleave the chromatin to the desired fragment size, thus reducing the resolution of the experiment.
Poor experimental repeatability: Because the conditions of excessive crosslinking are difficult to accurately control, the degree of crosslinking may vary from experiment to experiment, which will lead to poor repeatability of experimental results.
Sample loss: In the case of excessive crosslinking, complexes of protein and DNA may be lost in subsequent washing and purification steps because excessive crosslinking may enhance the binding force between these complexes.
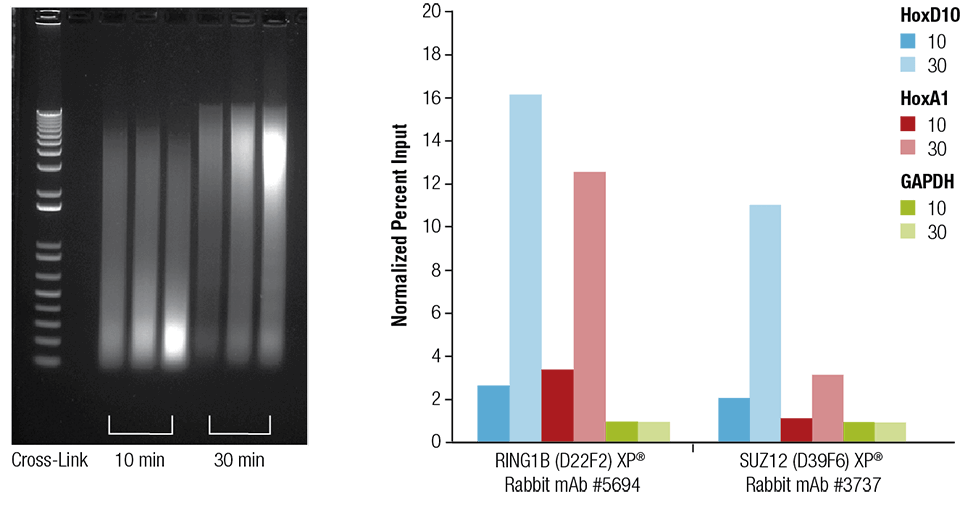
Fig. 3 Effect of crosslinking time
Mouse heart (H), brain (B) and liver (L) cells need to be cross-linked for 10 min or 30 min, as shown in the figure (left panel). Sonicated for 4 min after chromatin preparation, the enriched DNA was quantified by real-time PCR using the indicated gene primers (right panel). The amount of immunoprecipitated DNA in each sample is expressed as the ratio of normalized signal to negative site GAPDH (equal to 1).
* The picture comes from the Internet and is for learning reference only
8. Why fragment chromatin?
Improved antibody contact: Chromatin is a tightly bound complex of DNA and histones, usually found in the nucleus of cells. In ChIP experiments, specific antibodies need to be used to recognize and bind the protein of interest. Fragmenting the chromatin can increase the chances of an antibody contacting and binding to a protein of interest.
Facilitates immunoprecipitation: After chromatin fragmentation, DNA fragments are typically between 200-1000 base pairs in length. Such fragments facilitate antibody recognition and binding, and at the same time facilitate subsequent immunoprecipitation steps.
Improved resolution: By cutting chromatin into smaller fragments, ChIP experiments can provide higher resolution to more precisely locate the binding sites of proteins on the genome.
Reduction of background signal: Appropriate chromatin fragmentation helps reduce non-specific background signal, as larger chromatin fragments may contain multiple non-target binding sites, resulting in non-specific immunoprecipitation.
Easy DNA purification and analysis: Smaller DNA fragments are easier to separate from proteins, facilitating subsequent DNA purification and analysis, such as PCR, sequencing, or microarray analysis.
Improved experimental efficiency: Proper chromatin fragmentation can improve the efficiency of ChIP experiments because smaller fragments are more easily processed and analyzed, thereby reducing the complexity and time of experimental manipulation.
9. What is the difference between ultrasonic method and enzymatic hydrolysis?
Ultrasound: capable of producing DNA fragments of random size, suitable for experiments requiring assessment of histones and histone modifications, as these are enriching and stabilizing components of chromatin. The ultrasound method is suitable for X-ChIP, i.e. chromatin after crosslinking. But it is necessary to optimize the sonication conditions to avoid excessive crushing. Sonication generates heat, which needs to be carried out under ice-cold conditions, and the generation of bubbles should be avoided (bubbles will cause local temperatures to be ineffective cooled, affecting efficiency and accuracy, and at the same time causing uneven shear forces). The consistency and repeatability of the experiment are poor compared with enzymatic hydrolysis method.
Enzymatic hydrolysis: Mild fragmentation of chromatin using enzymes such as micrococcal nuclease (MNase) is more suitable for evaluating the binding of transcription factors and cofactors, since these factors may not be stable in interaction with DNA. Enzymatic hydrolysis is more reproducible between experiments, and the DNA fragments produced are relatively consistent in size, but enzymatic digestion may not produce random sections of chromatin, but rather cuts certain regions with preference, possibly causing certain sites to be overexpressed or missed. In addition, the enzymatic operation still requires sonication to break the nuclear membrane and release chromatin after digestion with enzymes.
The commonly used chromatin fragmentation methods of X-ChIP are enzymatic hydrolysis (micrococcal nuclease MNase) and ultrasound, while N-ChIP relies more on enzymatic hydrolysis.
10. How to determine the degree of chromatin fragmentation?
Agarose gel electrophoresis is the most commonly used method to check the effect of chromatin fragmentation. Thorough MNase digestion will produce DNA fragments (mononucleosomes) containing 150 base pairs, and incomplete digestion can also produce DNA fragments containing 150-750 base pairs (mononucleosomes, binucleosomes, and trinucleosomes). Sonication yields numerous chromatin fragments of 150-1000 base pairs. The minimum amount of sonication required to obtain the desired DNA size and avoid unnecessary chromatin damage is determined.

Fig. 4 Enzymatically digested chromatin treated on agarose gel
Lane 1 shows underdigested chromatin. Lane 2 shows properly digested chromatin, while lane 3 shows over-digested chromatin.
* The picture comes from the Internet and is for learning reference only
RECOMMENDED
|
Item number |
Trade Name |
Specifications |
|
Chromatin co-immunoprecipitation (ChIP) kit |
22T |
|
|
abs50073 |
DNA Pull Down Kit (Plant) |
6T |
|
abs50074 |
DNA Pull Down Kit (Animal) |
6T |
|
Immunoprecipitation (IP/CoIP) kit (magnetic bead method) |
10T/50T |
|
|
Immuno (co) precipitation (IP/CoIP) kit |
50T |
|
|
RNA co-immunoprecipitation (RIP) kit |
6T |
|
|
RNA Pull Down Kit |
6T |
|
|
abs50082 |
GST Pull Down Kit |
6T |



